Laskennallinen kemia: menetelmät, sovellukset ja molekyylien ennustaminen
Laskennallinen kemia: menetelmät ja sovellukset — ennusta molekyylien rakenteet, ominaisuudet ja reaktiivisuus uusien lääkkeiden ja materiaalien suunnittelussa.
Laskennallinen kemia on kemian osa-alue, jossa käytetään tietotekniikkaa kemiallisten ongelmien ratkaisemiseen. Nämä ohjelmat laskevat molekyylien ja kiinteiden aineiden rakenteita ja ominaisuuksia. Laskennallinen kemia täydentää yleensä kemiallisista kokeista saatua tietoa. Sillä voidaan ennustaa kemiallisia ilmiöitä, joita ei ole vielä havaittu. Sitä käytetään laajalti uusien lääkkeiden ja materiaalien suunnittelussa.
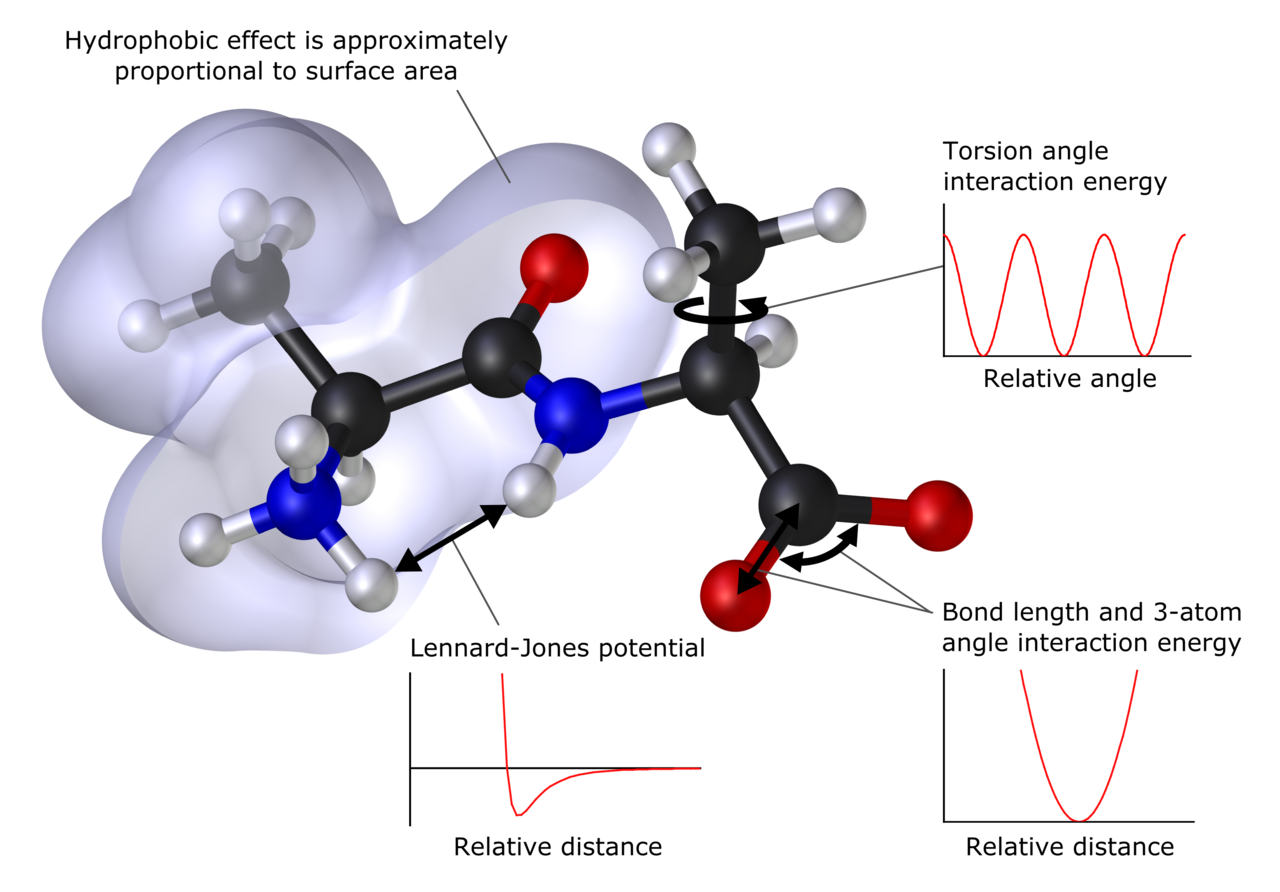
Laskennallisella kemialla voidaan ennustaa rakenne (eli molekyylin atomien odotetut sijainnit), absoluuttiset ja suhteelliset (vuorovaikutus)energiat, elektronisten varausten jakaumat, dipolit ja korkeammat monipolimomentit, värähtelytaajuudet, reaktiivisuus tai muut spektroskooppiset suureet sekä ristikkäisleikkaukset törmäyksissä muiden hiukkasten kanssa.
Laskennallisessa kemiassa tarkastellaan sekä staattisia että dynaamisia järjestelmiä. Kaikissa tapauksissa tutkittavan järjestelmän koon kasvaessa myös käytetty tietokoneaika ja muut resurssit (kuten muisti ja levytila) kasvavat. Järjestelmä voi olla yksittäinen molekyyli, molekyyliryhmä tai kiinteä aine. Laskennallisen kemian menetelmät vaihtelevat erittäin tarkoista hyvin likimääräisiin. Erittäin tarkat menetelmät ovat yleensä toteutettavissa vain pienille systeemeille.
Kuvagalleria
8 Kuvat




Keskeiset menetelmät
Laskennallisessa kemiassa käytetään useita eri lähestymistapoja, jotka eroavat tarkkuudeltaan, kustannuksiltaan ja soveltuvuudeltaan eri ongelmiin. Tyypilliset menetelmäluokat ovat:
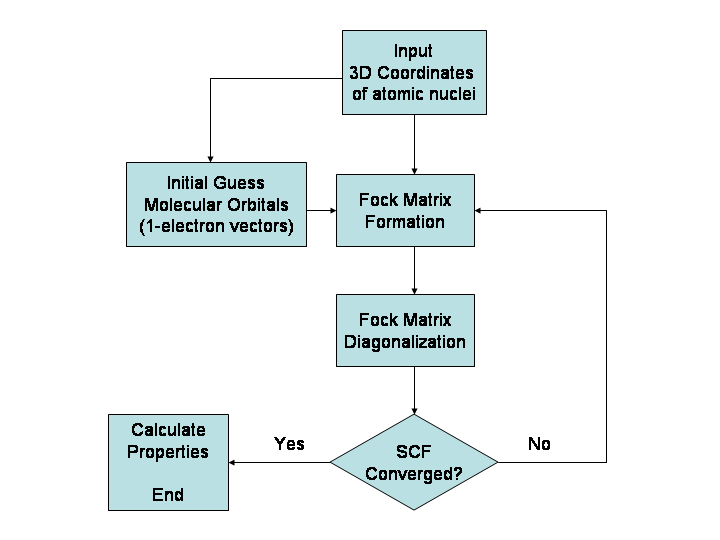
- Ab initio -menetelmät (ensimmäisperiaatteet): ratkaisevat elektronirakenteen kvanttimekaanisesti ilman empiirisiä parametreja. Esimerkkejä: Hartree–Fock (HF), MP2, CCSD(T). Näillä saavutetaan korkea tarkkuus pienille ja keskisuurille molekyyleille, mutta ne skaalautuvat huonosti koon kasvaessa.
- Tiheysfunktionaaliteoria (DFT): tasapainottaa tarkkuuden ja laskennallisen kustannuksen hyvin ja on yleisesti käytetty laajassa sovellusvalikoimassa (molekyylit, pinnat, kiinteät aineet). Funktionaalin valinta (esim. B3LYP, PBE, ωB97X-D) vaikuttaa merkittävästi tuloksiin.
- Semi-empiriset menetelmät: yksinkertaistavat kvanttimekaniikkaa ja käyttävät parametrisoituja kaavoja (esim. PM6, DFTB) — nopeita ja käyttökelpoisia suurilla järjestelmillä, mutta vähemmän yleistettävissä tarkkuudeltaan.
- Molekyylimekaniikka ja voimalaitokset (force fields): klassinen kuvaus atomien vuorovaikutuksista (esim. AMBER, CHARMM, OPLS). Soveltuu erittäin suurille järjestelmille (biomolekyylit, polymeerit) ja dynaamiseen simulointiin (molekyylidynamiikka).
- Dynaamiset menetelmät: molekyylidynamiikka (MD) ja Monte Carlo (MC) kuvaavat ajan kehitystä ja termodynaamisia ominaisuuksia; voivat käyttää sekä kvantti- että klassisia potentiaaleja (ab initio MD, QM/MM).
Sovellukset
Laskennallinen kemia tukee monia kemian ja materiaalitieteen alueita:
- Lääkesuunnittelu: rakenne–aktiivisuussuhteiden (SAR) analyysi, molekyylidokkaus, virtuaalinen seulonta, ligandin sitoutumisen ennustus ja ADMET-ominaisuuksien alustava arviointi.
- Materiaalitutkimus: akku- ja katalysaattorimateriaalien suunnittelu, puolijohteiden ja pintojen sähköiset ominaisuudet, mekaaniset ominaisuudet atomitasolla.
- Reaktiomekaniikka: siirtymätilojen etsintä, potentiaalienergiakäyrät, reaktioreittien ja -nopeuksien laskeminen.
- Spektroskopia: IR-, Raman-, NMR- ja UV/vis-spektrien laskenta ja tulkinta sekä rinnakkainen kokeellinen ja laskennallinen analyysi.
- Suuret tietoaineistot ja korkean läpimenon seulonta: virtuaalinen kandidivahti uusien yhdisteiden löytämiseen ennen kokeellista testausta.
Menetelmän valinta ja käytännön seikat
Oikean menetelmän valinta riippuu tavoitteesta: tarvittavasta tarkkuudesta, järjestelmän koosta ja resursseista. Tärkeimpiä huomioitavia seikkoja:
- Mittakaava ja skaalaus: ab initio -menetelmät skaalautuvat huonosti (esim. CCSD(T) ~ N^7), DFT on usein parempi kompromissi, ja molekyylimekaniikka mahdollistaa miljoonien atomien simuloinnin.
- Basis-setit ja parametrien valinta: kvanttilaskuissa käytetään basis-setejä (esim. 6-31G*, cc-pVTZ), joiden laatu vaikuttaa tuloksiin. Myös funktionaalin ja dispersiokorjausten valinta DFT:ssä on kriittinen.
- Ympäristömallit: liuottimen vaikutus voidaan huomioida implisiittisillä malleilla (PCM, COSMO) tai eksplisiittisellä veden mallinnuksella.
- Virheet ja validointi: laskennalliset ennusteet tulee aina validoida kokeellista dataa vastaan tai korkeimman tason laskelmia vasten; huomioon otettavia virheitä ovat mm. basis set superposition error (BSSE) ja funktionaalien systemaattiset virheet.
- Reproducibility: kirjaa ohjelmistoversiot, parametrit, lähtö- ja syöteasetukset sekä satunnaissiementen arvot, jotta tulokset voidaan toistaa.
Ohjelmistot ja laskentaresurssit
Yleisimmät laskennallisen kemian ohjelmistot kattavat sekä sähköisen rakenteen laskennan että dynaamiset simuloinnit. Esimerkkejä:
- Kvanttimekaaniset: Gaussian, ORCA, NWChem, Q-Chem, PSI4
- Kiinteän aineen DFT: VASP, Quantum ESPRESSO, CP2K
- Molekyylimekaniikka ja MD: GROMACS, AMBER, CHARMM, LAMMPS
- Uudemmat lähestymistavat: koneoppimispohjaiset potentiaalit (esim. ANI, SchNet), GPU-pohjaiset laskelmat ja pilvipalvelut tarjoavat lisää laskentakykyä.
Nykyiset trendit ja tulevaisuus
- Koneoppiminen ja tekoäly: ML-mallit nopeuttavat energialaskelmia, optimoivat potentiaaleja ja tukevat suurtehokasta seulontaa.
- GPU-kiihdytys: nopeuttaa erityisesti molekyylidynamiikkaa ja joitakin kvanttimekaniikan osia.
- Hybridimenetelmät: QM/MM-tekniikat yhdistävät kvanttikemian ja klassisen kuvaamisen tarjoten tarkkuutta aktiivisissa kohdissa ja tehokkuutta suurissa järjestelmissä.
- High-throughput -lähestymistavat: automatisoitu laskentaputki ja tietokannat (Materials Project, CSD) mahdollistavat suuret aineistot materiaalien löytämiseksi.
Rajoitukset ja eettiset näkökulmat
Laskennallinen kemia tarjoaa voimakkaita työkaluja, mutta sillä on rajoituksensa: kaikki tulokset perustuvat malleihin ja approksimaatioihin, joten virheiden mahdollisuus on aina olemassa. On tärkeää ilmoittaa epävarmuudet ja varmistaa, että laskelmien johtopäätökset eivät korvaa kokeellista varmistusta, vaan ohjaavat ja täydentävät sitä. Lisäksi avoimuus, toistettavuus ja datan jakaminen parantavat tutkimuksen luotettavuutta.
Yhteenveto
Laskennallinen kemia on monipuolinen ja kehittyvä ala, joka yhdistää teoreettisen kemian, algoritmit ja tehokkaan laskentakapasiteetin. Se tarjoaa ennusteita ja syvempää ymmärrystä atomitason ilmiöihin, nopeuttaa uusien aineiden löytämistä ja tukee kokeellista työtä. Menetelmien oikein valitseminen, tulosten validointi ja selkeä dokumentointi ovat avainasemassa luotettavien ja käyttökelpoisten tulosten saamiseksi.

Aiheeseen liittyvät sivut
- Bioinformatiikka
- Tilastollinen mekaniikka
Kysymyksiä ja vastauksia
K: Mitä on laskennallinen kemia?
V: Laskennallinen kemia on kemian osa-alue, jossa käytetään tietojenkäsittelyä kemiallisten ongelmien ratkaisemisessa. Sen avulla voidaan laskea molekyylien ja kiinteiden aineiden rakenteita ja ominaisuuksia, ennustaa kemiallisia ilmiöitä, joita ei ole vielä havaittu, ja suunnitella uusia lääkkeitä ja materiaaleja.
K: Millaisia järjestelmiä laskennallinen kemia tarkastelee?
V: Laskennallinen kemia tarkastelee sekä staattisia että dynaamisia järjestelmiä. Systeemi voi olla yksittäinen molekyyli, molekyyliryhmä tai kiinteä aine.
K: Millaista tietoa laskennallinen kemia voi tarjota?
V: Laskennallinen kemia voi tarjota tietoa esimerkiksi rakenteesta (atomien sijainnit), absoluuttisista ja suhteellisista energioista, elektronisten varausten jakaumista, dipoleista ja korkeammista monipolimomenteista, värähtelytaajuuksista, reaktiivisuudesta tai muista spektroskooppisista suureista sekä ristikkäisleikkauksista törmätessä muiden hiukkasten kanssa.
K: Kuinka tarkkoja laskennallisessa kemiassa käytetyt menetelmät ovat?
V: Laskennallisessa kemiassa käytettävien menetelmien tarkkuus vaihtelee erittäin tarkasta hyvin likimääräiseen. Erittäin tarkat menetelmät ovat yleensä mahdollisia vain pienille systeemeille.
K: Miten laskennallinen kemia täydentää kokeellista tietoa?
V: Laskennallinen kemia täydentää yleensä kemiallisista kokeista saatua tietoa. Sen avulla voidaan ennustaa tuloksia, joita ei ole vielä havaittu kokeellisesti.
K: Vaikuttaako tutkittavan järjestelmän koko siihen, kuinka paljon tietokoneaikaa tarvitaan?
V: Kyllä - tutkittavan järjestelmän koon kasvaessa myös analyysiin tarvittava tietokoneaika sekä resurssit, kuten muisti ja tallennukseen tarvittava levytila, kasvavat.
Aiheeseen liittyvät artikkelit
Tekijä
AlegsaOnline.com Laskennallinen kemia: menetelmät, sovellukset ja molekyylien ennustaminen Leandro Alegsa
URL: https://fi.alegsaonline.com/art/22297