Osteogenesis imperfecta – haurasluusairaus, oireet, tyypit ja ennuste
Osteogenesis imperfecta haurasluusairaus, geneettiset syyt, oireet ja tyypit 1-4, murtumariski, kollageenivika, kuulon heikkeneminen ja ennuste sekä elämänlaatuun vaikuttavat seikat
Osteogenesis imperfecta on geneettinen sairaus, jota kutsutaan yleisesti hauraan luun sairaudeksi. Se johtuu virheestä erityisesti kollageeni‑tyypin I muodostavissa geeneissä, jolloin luiden kollageenisäikeet heikentyvät. Näin luut eivät kestä normaalia kuormitusta ja murtuvat helpommin. Taudin ensimmäisiä kuvaajia olivat 1800‑luvulla toimineet tutkijat, ja sittemmin sen perinnöllinen luonne sekä keskeiset geenivirheet on tunnistettu.
Kuvagalleria
10 Kuvat







Oireet
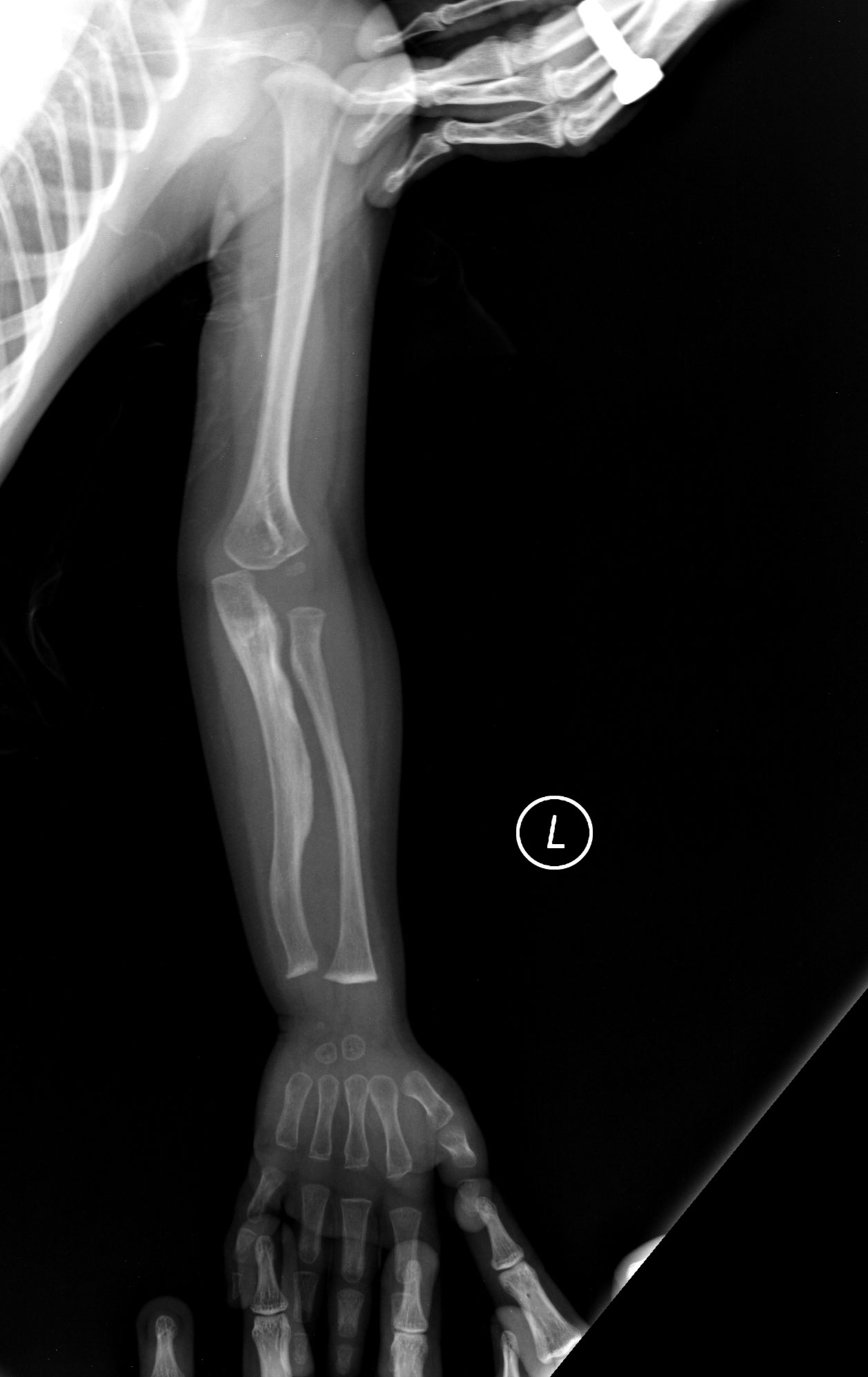
- Murtumat: toistuvat luunmurtumat pienienkin vammojen seurauksena.
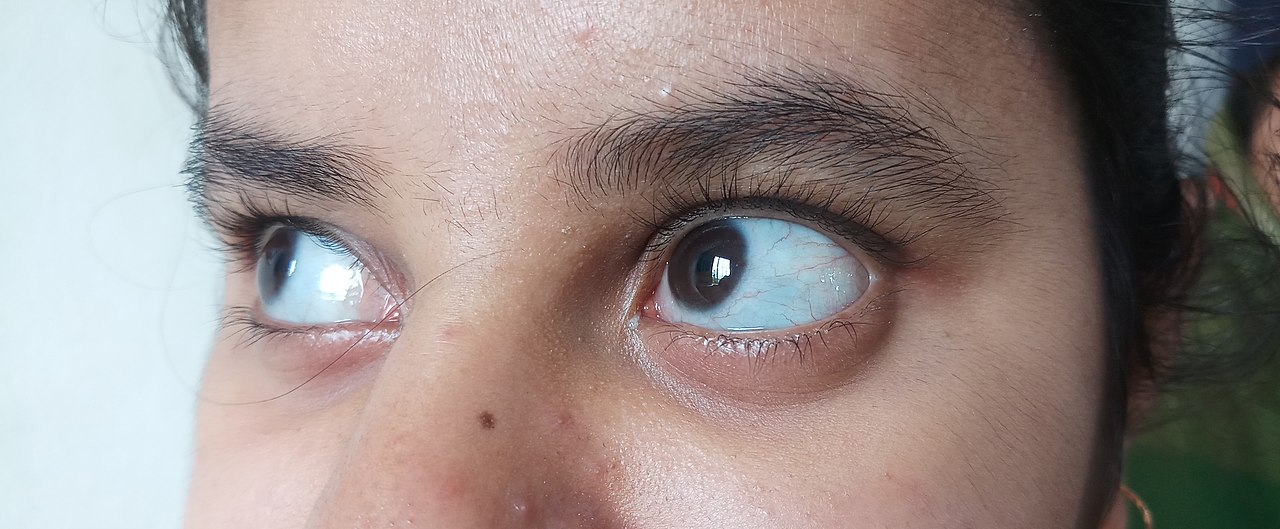
- Sinertävät silmän valkuaiset (sinertävät sklerat): tyypillinen löydös, johtuu ohuesta skleraasta.
- Lyhyt kasvu ja vartalon muodon muutokset: vaikeissa muodoissa voi esiintyä kasvun hidastumista ja epämuodostumia.
- Hammashuolet: dentinogenesis imperfecta eli kiilteen ja hammasluun kehityshäiriöt, herkät ja kuluneet hampaat.
- Kuulon heikkeneminen: erityisesti aikuisilla tyypeissä I–IV kuuluva ongelma.
- Rintakehän ja hengityselinten ongelmat: vaikeammissa muodoissa voivat johtaa hengitysvaikeuksiin.
- Luuston muutosongelmat: skolioosi (selkärangan vääntyminen), nivelten liikkeiden rajoitukset.
Syyt ja periytyminen
Suurin osa tapauksista johtuu mutaatioista geeneissä COL1A1 tai COL1A2, jotka koodaavat kollageeni‑tyypin I:n rakennusosia. Periytyminen on usein autosomaalinen dominantti, eli riittää, että vain toisella vanhemmista on virheellinen geeni, jotta lapsi voi periä sairauden. On kuitenkin olemassa myös harvinaisempia periytymismalleja ja muitakin mukana olevia geenejä.
Tyyppijako
Perinteisesti Osteogenesis imperfecta on jaettu neljään päätyyppiin (Sillence‑luokitus), joiden vaikeusaste vaihtelee:
- Tyyppi I: yleisin ja yleensä lievin muoto. Murtumia esiintyy, mutta elinikä on usein normaali. Usein myös sinertävät sklerat ja dentinogenesis imperfecta.
- Tyyppi II: vakavin muoto, usein vasta syntymän yhteydessä tai varhaisina elinviikkoina hengitys- tai muiden ongelmien vuoksi kuolemaan johtava.
- Tyyppi III: vakava, usein progressiivinen muoto, johon liittyy runsas määrä murtumia, merkittävä lyhytkasvuisuus ja luuston vääristymiä. Potilailla voi olla yli 100 murtumaa ennen murrosikää.
- Tyyppi IV: keskivaikea tai haittava muoto, joka sijoittuu tyypin I ja III välimaastoon; murtumia ja dentinogenesis imperfectaa voi esiintyä.
Uudemmat luokitukset tunnistavat useampia tyyppejä (V, VI jne.), mutta kliinisesti yleisin lähestymistapa on edelleen arvioida vakavuusaste ja yksilölliset oireet.
Diagnoosi
- Kliininen arvio: sairaushistoria (toistuvat murtumat), fyysiset löydökset kuten sinertävät sklerat ja kasvun poikkeavuudet.
- Röntgenkuvat ja muu kuvantaminen luuston arvioimiseksi.
- Genetinen testaus: varmistaa mutaation paikantamisen (esim. COL1A1/COL1A2).
- Laboratoriotutkimukset ja erikoisalojen (ortopedi, lastentauti, endokrinologi, hammaslääkäri, audiologi) konsultaatioita usein tarvitaan.
Hoito ja seuranta
Parannuskeinoa ei ole, mutta oireita pyritään ehkäisemään ja hoitamaan monialaisesti. Hoidon tavoitteena on vähentää murtumia, ylläpitää toimintakykyä ja hallita kipua.
- Bisfosfonaattiterapia: luun tiheyden lisäämiseksi ja murtumariskin vähentämiseksi, käytössä erityisesti lapsilla ja vaikeammissa tapauksissa.
- Ortopedinen hoito: murtumien asianmukainen hoito, kirurgiset toimenpiteet kuten intramedullaariset kepit (rodding) luiden muodon korjaukseen ja tukemiseen.
- Fysioterapia ja kuntoutus: lihasvoiman ylläpito, liikkeiden hallinta ja apuvälineet liikkumisen tukemiseksi.
- Hammaslääketiede: säännöllinen seuranta ja hoito dentinogenesis imperfectaa varten.
- Kuulo: kuulontutkimukset ja apuvälineet kuten kuulokojeet tarvittaessa.
- Hengitystoiminnan seuranta: etenkin vaikeissa muodoissa tärkeää.
Ennuste
Ennuste riippuu suuresti OI‑tyypistä ja yksilöllisestä vakavuudesta. Monet tyypin I potilaat elävät normaalia elämää ja ovat pitkään aktiivisia, kun taas tyyppi II voi olla vastasyntyneellä hengenvaarallinen. Tyypit III–IV voivat aiheuttaa pitkäaikaisia vammoja ja toimintakyvyn rajoituksia, mutta elinikä voi silti olla pitkälle aikuisiän ulottuva olosuhteista ja hoidon onnistumisesta riippuen.
Neuvonta ja ehkäisy
Genetinen neuvonta on tärkeää perheille, joissa OI on todettu. Genetinen testaus mahdollistaa kantajuuden selvittämisen, perinnöllisyysriskien arvioinnin sekä mahdollisuuden prenaatala- ja esivalintadiagnostiikkaan (PGD) tilanteen mukaan.
Elämänhallinta ja käytännön vinkkejä
- Suojele luustoa yksilöllisillä keinoilla: liikunnan räätälöinti, turvallinen ympäristö ja asianmukaiset apuvälineet.
- Pidä säännölliset kontrollit hammaslääkärille, kuulontutkimuksiin ja ortopedille.
- Moni tarvitsee moniammatillisen tiimin tukea: lastentauti‑/aikuislääkäri, ortopedi, fysioterapeutti, hammaslääkäri ja genetiikan asiantuntija.
Jos epäilet Osteogenesis imperfectaa itselläsi tai lapsellasi, ota yhteyttä hoitavaan lääkäriin tai perinnöllisyysneuvontaan jatkotutkimuksia ja ohjausta varten. Oikeanlaisella seurannalla ja hoidolla monen potilaan elämänlaatua voidaan merkittävästi parantaa.
Oireet
OI:n lievempiä oireita voivat olla:
- helposti murtuvat luut
- löysät nivelet
- alhainen lihasjänteys
- sinistä, violettia tai harmaata väriä silmien normaalisti valkoisessa osassa.
- kasvojen kolmiomainen muoto
- taipumus kehittää skolioosi
- hauraat hampaat
OI:llä on monia muita vakavia ja kuolemaan johtavia oireita, kuten hengitystieongelmia ja luun epämuodostumia.
Demografiset tiedot
OI:tä esiintyy yhtä lailla sekä miehillä että naisilla, ja se voi vaikuttaa kaikkiin etnisiin ryhmiin. OI syntyy kohdussa, eikä siihen ole parannuskeinoa. Se havaitaan yleensä silloin, kun joku murtuu paljon luita; hänelle voidaan tehdä DNA-testi OI:n toteamiseksi. Sitä esiintyy yhdellä 20 000:sta syntyneestä.
Kysymyksiä ja vastauksia
K: Mikä on osteogenesis imperfecta?
V: Osteogenesis imperfecta on geneettinen sairaus, jota kutsutaan yleisesti hauraan luun sairaudeksi. Se heikentää tai tuhoaa kollageenisauvaa, joka antaa luun lujuuden, ja johtaa siihen, että luut murtuvat herkemmin.
K: Miten osteogenesis imperfecta periytyy?
V: Osteogenesis imperfecta on yleensä autosomaalinen dominantti sairaus, mikä tarkoittaa, että henkilö voi sairastua siihen, jos vain toisella vanhemmista on epänormaali geeni.
K: Kuka tunnisti ensimmäisenä osteogenesis imperfectan?
V: Vrolik tunnisti ensimmäisenä osteogenesis imperfectan vuonna 1849.
K: Onko osteogenesis imperfectaan olemassa parannuskeinoa?
V: Valitettavasti osteogenesis imperfectaan ei ole parannuskeinoa.
K: Mitkä ovat osteogenesis imperfectan neljä tyyppiä?
V: Osteogenesis imperfectan neljä tyyppiä ovat tyyppi yksi, tyyppi kaksi, tyyppi kolme ja tyyppi neljä.
K: Mitkä ovat tyypin kolme osteogenesis imperfecta oireet?
V: Ihmisillä, joilla on osteogenesis imperfecta -tyypin kolme, voi olla yli 100 murtumaa ennen murrosikää. Heidän silmiinsä kehittyy usein violetti, sininen tai harmaa sävy, ja tätä tapausta sairastavilla on usein myös kuulon heikkeneminen.
K: Kuinka yleistä kuulon heikkeneminen on osteogenesis imperfectaa sairastavilla?
V: 50 prosentilla ihmisistä, joilla on osteogenesis imperfecta, on kuulon heikkeneminen aikuiseksi tullessaan.
Aiheeseen liittyvät artikkelit
Tekijä
AlegsaOnline.com Osteogenesis imperfecta – haurasluusairaus, oireet, tyypit ja ennuste Leandro Alegsa
URL: https://fi.alegsaonline.com/art/73422
Lähteet
- oif.org : "Osteogenesis Imperfecta Foundation:"
- nlm.nih.gov : "Autosomal dominant: MedlinePlus Medical Encyclopedia"
- oif.org : "Osteogenesis Imperfecta Foundation: Understanding Bone Structure"