Sykloadditio – määritelmä, mekanismi ja esimerkit kuten Diels–Alder
Sykloadditio pähkinänkuoressa: määritelmä, perisyklinen mekanismi ja esimerkit kuten Diels–Alder [4+2] ja 1,3-dipolaarinen [3+2] reaktioiden kulku, stereokemia ja sovellukset
Sykloaditio on kemiallinen reaktio, jossa kahden tai useamman tyydyttymättömän reagoijan kaksois- tai kolmoissidoksia “kulutetaan” ja ne korvautuvat uudella rengasrakenteella. Se on perisyklinen kemiallinen reaktio, joka etenee yleensä yhdessä, samanaikaisessa siirtymätilassa: tyypillisesti kaksi tai useampia tyydyttymättömiä molekyylejä (tai saman molekyylin osia) yhdistyy muodostaen syklisen adduktin, ja sidosten kokonaislukumäärä “yksinkertaistuu” nettomääräisesti. Kyseessä on siis syklisoitumisreaktio, jossa syntyy uusi rengas atomeista ilman välivälituotteita.
Sykloadditiot nimetään yhdistyvien molekyylien π-komponenttien atomimäärän mukaan hakasuljenotaatiolla [i + j]. Tällöin Diels-Alder-reaktio on [4 + 2] -sykloadditio ja 1,3-dipolaarinen sykloadditio [3 + 2] -sykloadditio. Tämäntyyppinen reaktio on luonteeltaan pooliton additioreaktio.
Kuvagalleria
6 Kuvat





Mekanismi ja orbitaalisäännöt
Sykloadditiot ovat tavallisesti yhdessä (concerted) eteneviä: sidokset katkeavat ja muodostuvat samanaikaisesti syklisessä elektroninsiirtosilmukassa. Reaktion kulku selittyy kahdella toisiaan täydentävällä tavalla:
- Orbitaalien symmetria (Woodward–Hoffmannin säännöt): Lämpöolosuhteissa sallitut sykloadditiot etenevät niin, että orbitaalit kohtaavat symmetriaa säilyttäen. Esimerkiksi [4 + 2] on lämpöisesti sallittu (suprafasiaalinen–suprafasiaalinen), kun taas yksinkertainen [2 + 2] on lämpöisesti kielletty mutta valokemiallisesti sallittu.
- Rajamolekyyliorbitaalit (FMO): Reaktionopeus määräytyy pääasiassa reagoijien HOMO–LUMO-yhteensopivuudesta. Normaalissa elektronikysynnässä dieni on elektronirikas (esim. -OR, -alkyyli) ja dienofiili elektroniköyhä (esim. -CN, -COR, -COOR). Käänteisessä elektronikysynnässä roolit vaihtuvat.
Diels–Alder ([4 + 2])
- Perusreaktio: 1,3-butadieeni + alkeeni (esim. eteeni) → sykloheks-1-eeni. Reaktio on stereospesifinen: dienofiilin cis-/trans-geometria säilyy rengastuotteessa.
- Endo-/exo-valikoivuus: Elektronia vetäviä π-ryhmiä sisältävien dienofiilien kanssa endo-tuote on usein kinetisesti suosittu (endo-sääntö), koska toissijaiset orbitaali-interaktiot stabiloivat siirtymätilaa.
- Regioselektiivisyys: Epäsymmetristen reaktanttien kanssa substituenttien ohjaus johtaa ennustettavaan orto-/para-analogiaan; FMO-analyysi auttaa tuotteiden suhteiden ennustuksessa.
- Hetero-Diels–Alder: Kun toinen komponentti sisältää heteroatomin (esim. C=O, C=N), saadaan heterosyklisiä renkaita, kuten dihydropyraneja tai dihydropyridiinejä.
- Intramolekulaarinen DA: Sama molekyyli sisältää sekä dienin että dienofiilin; muodostuu tiiviitä, usein polytsyklisten luonnonaineiden runkoja.
- Käänteisreaktio (retro-DA): Lämmittäminen voi pilkkoa renkaan takaisin lähtökomponentteihin; tätä hyödynnetään suojaryhmästrategioissa ja aromaattisten fragmenttien vapautuksessa.
- Katalyysi ja olosuhteet: Lewis-hapot (esim. BF3, AlCl3, TiCl4) aktivoivat dienofiilin ja kiihdyttävät reaktiota sekä parantavat selektiivisyyttä. Korkea paine (negatiivinen aktivaatiotilavuus) ja kohtalainen lämpö nopeuttavat; liuottimen polarisoituvuus voi lisätä nopeutta.
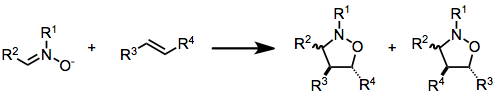
1,3-dipolaariset sykloadditiot ([3 + 2])
- Tyypilliset 1,3-dipolit: Orgaaniset atsidit (N3−), nitronit, nitrilioksidit ja atsometiiniylidit.
- Tuotteet: Viisijäseniset heterosyklit, kuten 1,2,3-triatsolit, isoksatsolidit ja pyrrolidiinijohdannaiset.
- “Click”-kemia: Kupari(I)-katalysoitu atsidi–alkyyni-sykloadditio (CuAAC) tuottaa selektiivisesti 1,4-disubstituoituja 1,2,3-triatsoleja; reaktio on nopea, siedettää vettä ja monia funktionaaliryhmiä ja on bioortogonaalinen.
- Stereokemia ja regio-ohjaus: Substituenttivaikutukset ja mahdollinen metallikatalyysi ohjaavat regioselektiivisyyttä; asymmetriset katalyytit mahdollistavat korkean enantioselektiivisyyden.
Muita sykloadditioita ja erityistapauksia
- [2 + 2] -sykloadditiot: Kaksi alkeenia muodostaa syklobutaanin tyypillisesti valokemiallisesti. Karbonyylin ja alkeenin [2 + 2] tuottaa oksetaneja (klassinen valokemiallinen Paternò–Büchi-tyyppinen lisäys).
- [4 + 3] ja [5 + 2]: Antavat seitsemän- ja seitsenjäsenisiä renkaita esimerkiksi oksiallyylivälitteisesti ([4 + 3]) tai koordinaatiokatalyysillä ([5 + 2]).
- Keletrooppiset lisäykset: Kaksi uutta sidosta muodostuu samaan atomiin (esim. SO2:n additio; käänteisesti SO2:n eliminoituminen retro-DA-tyyppisesti).
Selektiivisyys ja suunnittelu
- Stereospesifisyys: Reagoijien cis/trans- ja E/Z-geometria heijastuu suoraan tuotteeseen; uusia kiraalikeskuksia muodostuu määritellysti.
- Regioselektiivisyys: Ennustettavissa FMO-analyysillä ja substituenttien ohjausvaikutuksilla; usein voidaan kääntää lisäämällä aktivoivia/poistavia ryhmiä tai vaihtamalla katalyyttiä.
- Asymmetrinen induktio: Kiraaliset Lewis-happokatalyytit ja orgaaniset katalyytit mahdollistavat korkean enantioselektiivisyyden.
Käytännön vinkkejä ja olosuhteiden optimointi
- Lämpötila: Kohtalainen kuumennus (esim. 25–120 °C) nopeuttaa ilman sivureaktioita; liian korkea lämpö voi suosia retro-reaktiota.
- Paine: Korkea paine (kymmenistä satoihin MPa) voi merkittävästi nopeuttaa ja parantaa saantoa erityisesti Diels–Alderissa.
- Liuotin: Polaariset, heikosti nukleofiiliset liuottimet (esim. dikloorimetaani, nitrometaani) ovat usein hyviä; vesi voi nopeuttaa joitakin Diels–Alder-reaktioita hydrofobisen vaikutuksen kautta.
- Suunnittelu: Dienten on oltava s-cis-konformaatiossa; steriset esteet ja konjugaatio vaikuttavat voimakkaasti reaktionopeuteen.
Sovellukset
- Luonnonaine- ja lääkesynteesi: Nopea renkaiden rakentaminen ja useiden kiraalikeskusten luonti yhdessä vaiheessa.
- Materiaalikemia: Verkottuminen, itsekorjautuvat materiaalit (esim. lämpöreversiibelit retro-DA-sidokset), valokemialliset kuvioinnit.
- Kemiallinen biologia: Bioortogonaaliset merkinnät (esim. triatsolien muodostus), proteiinien ja biomolekyylien hellävarainen konjugointi.
Reaktiomekanismi
Lämpö voi saada kaksoissidokset muodostamaan renkaan. Lämpösykladitiossa on yleensä (4n + 2) π-elektronia, jotka osallistuvat lähtöaineeseen jollakin kokonaisluvulla n. Orbitaalisymmetrian vuoksi useimmat sykladitiot ovat suprafaasisia-suprafaasisia. Harvoin ne ovat antarafacial-antarafacial. On olemassa muutamia esimerkkejä termisistä sykloadditioneista, joissa on 4n π-elektronia (esimerkiksi [2 + 2]-sykloaddition). Nämä etenevät suprafacial-antarafacial-mielessä. Esimerkiksi keteenin dimerisaatiossa on ortogonaalinen joukko p-orbitaaleja. Näiden p-orbitaalien ansiosta reaktio voi edetä ristikkäisen siirtymätilan avulla.
Valo voi myös saada kaksoissidokset muodostamaan renkaan. Sykloadditiot, joihin 4n π -elektronit osallistuvat, voivat myös tapahtua valokemiallisen aktivoinnin seurauksena. Tällöin yksi komponentti saa elektronin siirtymään korkeimmin miehitetystä molekyyliorbitaalista (HOMO) (π-sidos) alimpaan miehittämättömään molekyyliorbitaaliin (LUMO) (π*-antisidos). Kun elektroni on siirtynyt korkeampaan orbitaaliin, orbitaalien symmetrian ansiosta reaktio voi edetä suprafacial-suprafacial-periaatteella. Esimerkki on DeMayo-reaktio. Toinen esimerkki on alla esitetty, kanelihapon fotokemiallinen dimerisaatio.
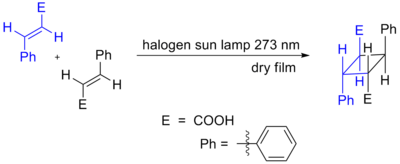
On huomattava, että kaikki fotokemialliset (2+2)-syklisaatiot eivät ole sykloadditioita; joidenkin tiedetään toimivan radikaalimekanismeilla.
Jotkin sykloadditiot toimivat π-sidosten sijasta kireiden syklopropaanirenkaiden kautta, koska niillä on merkittävä π-luonne. Esimerkiksi Diels-Alder-reaktion analogi on kvadrisyklaani-DMAD-reaktio:
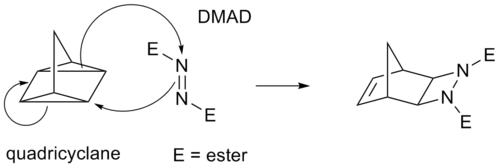
(i+j+...) sykladition merkinnöissä i ja j viittaavat sykladitioon osallistuvien atomien lukumäärään. Tässä merkintätavassa Diels-Alder-reaktio on (4+2)-sykladition ja 1,3-dipolaarinen additio, kuten otsonolyysin ensimmäinen vaihe, on (3+2)-sykladition. Tässä merkinnässä käytetään sulkuja. IUPACin suosima merkintätapa [i+j+...] laskee kuitenkin elektronit eikä atomeja. Siinä käytetään hakasulkeita. Tässä merkintätavassa Diels-Alder-reaktio ja dipolaarinen reaktio muuttuvat molemmat [4+2]sykladitioniksi. Norbornadieenin ja aktivoidun alkiinin välinen reaktio on [2+2+2]sykladition.
Sykladition tyypit
Diels-Alder-reaktiot
Diels-Alderin reaktio on [4+2]sykliliitosreaktio.
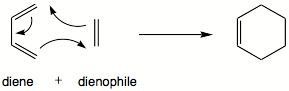
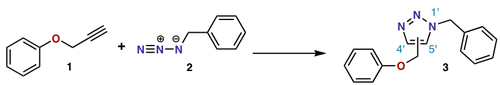
Huisgenin sykloadditiot
Huisgenin sykloadditioreaktio on [2+3]sykloadditioreaktio.
Nitroni-olefiinisykliliitos
Nitroni-olefiinisykliliitos on [3+2]sykliliitos.
Muodolliset sykladitiot
Sykladitioilla on usein metallikatalysoituja ja vaiheittaisia radikaalianalogeja, mutta nämä eivät varsinaisesti ole perisyklisiä reaktioita. Kun sykloadditiossa on mukana varattuja tai radikaaleja välituotteita tai kun sykloaddition tulos löytyy reaktiovaiheiden sarjasta, niitä kutsutaan joskus muodollisiksi sykloadditioiksi erona todellisiin perisyklisiin sykloadditioihin.
Yksi esimerkki n-butyylitiumin katalysoimasta muodollisesta [3+3]syklisen enonin ja enamiinin välisestä syklisestä [3+3]sykliliitoksesta on Stork-enamiini/1,2-additiokaskadireaktio:
![Intermolecular Formal [3+3] Cycloaddition Reaction](https://www.alegsaonline.com/image/600px-3%2B3-cycloaddition.svg.png)
Kysymyksiä ja vastauksia
Kysymys: Mikä on sykloaddition?
A: Sykloaditio on kemiallinen reaktio sellaisten reaktanttien välillä, joilla on kaksoissidoksia, jotka korvautuvat rengasrakenteella.
K: Minkä tyyppinen kemiallinen reaktio on sykloaddition?
V: Sykloaddition on perisyklinen kemiallinen reaktio, jossa "kaksi tai useampi tyydyttymätön molekyyli (tai saman molekyylin osa) yhdistyy muodostaen syklisen adduktin, jossa sidosten moninaisuus vähenee nettomääräisesti".
Kysymys: Mitä sykladitioreaktio tekee?
V: Sykloaddition on syklisoitumisreaktio: se muodostaa atomeista uuden renkaan.
K: Miten sykloaditiot on nimetty?
V: Sykloaditiot nimetään yhteen liitettävien molekyylien peruskoon mukaan.
K: Mikä on Diels-Alderin reaktio?
V: Diels-Alderin reaktio on [4 + 2]-sykladition.
K: Mikä on 1,3-dipolaarinen sykloaddition?
V: 1,3-dipolaarinen sykloaddition on [3 + 2]-sykloaddition.
K: Minkä tyyppinen reaktio on sykloaditio?
V: Sykladition on pooliton additioreaktio.
Aiheeseen liittyvät artikkelit
Tekijä
AlegsaOnline.com Sykloadditio – määritelmä, mekanismi ja esimerkit kuten Diels–Alder Leandro Alegsa
URL: https://fi.alegsaonline.com/art/24866
Lähteet
- goldbook.iupac.org : Cycloaddition
- doi.org : 10.1021/ed083p940
- doi.org : 10.1002/anie.200603302
- pubmed.ncbi.nlm.nih.gov : 17146819