Molekyyliorbitaali – määritelmä, muodostuminen ja kvanttimekaniikka
Molekyyliorbitaali – selkeä määritelmä, muodostuminen ja kvanttimekaniikka. Opas MO-mallien, atomiorbitaalien ja elektronitiheyden ymmärtämiseen.
Kemiassa molekyyliorbitaali (tai MO) kuvaa sitä, mitä tapahtuu elektroneille, kun atomit liittyvät yhteen molekyylissä. MO on kvanttimekaaninen matemaattinen funktio, joka kuvaa elektronin aaltomaista käyttäytymistä ja jonka neliö kertoo todennäköisyyden löytää elektroni tietyltä alueelta molekyylin ympäriltä. Kemistit ja teoreettiset tutkijat käyttävät näitä funktioita ennustaakseen ja selittääkseen kemiallisia ja fysikaalisia ominaisuuksia, kuten sidosten muotoa, elektronitiheyttä, spektriä tai magneettisuutta.
Kuvagalleria
5 Kuvat
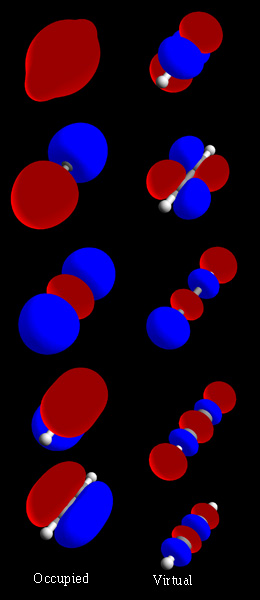



Kuinka molekyyliorbitaalit muodostuvat
Kemistit rakentavat usein matemaattisia molekyyliorbitaalimalleja yhdistämällä atomiorbitaaleja (LCAO‑menetelmä, linear combination of atomic orbitals). Kahdesta tai useammasta atomiorbitaalista muodostuu lineaarikombinaatioita, joista osa on konstruktivisia (sidosorbitaaleja) ja osa destruktivisia (antidi‑sidosorbitaaleja). Sidosorbitaalissa aaltofunktioilla on sama etumerkki ja elektronitiheys atomeja yhdistävällä alueella kasvaa, mikä stabiloi järjestelmää. Antisidoksisessa orbitaalissa esiintyy solmuja (nodal plane), ja yhteisenergia on korkeampi.
Orbitaalien energiajärjestys määräytyy alkuperäisten atomiorbitaalien energioista ja niiden lomittumisesta (overlap). Lähempänä toisiaan olevat ja hyvin lomittuvat orbitaalit muodostavat voimakkaamman sidoksen. Myös atomien hybridiorbitaaleja tai laajempia delokalisoituneita MOita voidaan käyttää kuvaamaan monimutkaisempia järjestelmiä.
Keskeiset käsitteet ja ominaisuudet
- Sidos- ja antisiidosorbitaalit: sidosorbitaalit (bonding) alentavat järjestelmän energiaa, antisiidosorbitaalit (antibonding, merkitty usein tähti‑merkinnällä kuten σ* tai π*) nostavat sitä.
- Sigma (σ) ja pi (π) orbitallit: σ‑orbitaalit ovat symmetrisiä atomitason akselin ympäri, π‑orbitaalit syntyvät sivittaisten päällekkäisyyksien kautta ja voivat johtaa delokalisoituneisiin π‑järjestelmiin (esim. alkeenit, aromaattiset yhdisteet).
- HOMO ja LUMO: korkein täytetty molekyyliorbitaali (Highest Occupied Molecular Orbital) ja matalin tyhjä (Lowest Unoccupied Molecular Orbital) määräävät usein kemiallisen reaktiivisuuden ja elektronisiirtymien todennäköisyyden.
- Elektronien täyttö: MO‑tasot täytetään Pauli‑periaatteen ja Aufbau‑periaatteen mukaisesti; Hundin sääntö koskee degeneraattisten orbitaalien elektronien sijoittumista.
- Todennäköisyys ja tiheys: MO on aaltofunktio ψ; elektronin löytämisen todennäköisyys on |ψ|2. Visualisoinnissa käytetään usein isopintatasoja (isosurfaces) näyttämään, missä elektronitiheys on merkittävää.
- Symmetria ja ryhmäteoria: molekyylin symmetria rajoittaa, mitkä atomiorbitaaleista voivat lomittua; ryhmäteoria auttaa MO‑nimityksissä ja energioiden ennustamisessa.
Esimerkkejä ja ilmiöitä
Yksinkertainen esimerkki on vetymolekyyli H2: kahden H‑1s‑orbitaalin yhdistyessä syntyy yksi sidosorbitaali (σ1s) ja yksi antisiidosorbitaali (σ1s*). Kaksi elektronia täyttävät sidosorbitaalin ja muodostavat yksinkertaisen kovalenttisen sidoksen. Toisaalta happimolekyyli O2 selittyy MO‑teorialla siten, että siinä on kaksi paritonta elektronia π*‑antisiidosorbitaaleissa, mikä selittää O2:n paramagnetisuuden — ilmiö, jota yksinkertainen sidosteoria ei kuvaa yhtä sujuvasti.
Laajemmissa π‑järjestelmissä, kuten bentseenissä, MO‑teoria selittää elektronien delokalisaation ja aromaattisuuden: π‑elektronit jakautuvat koko renkaan yli ja muodostavat energialtaan eriytyneitä delokalisoituneita orbitaaleja.
Kvanttimekaniikka, laskennalliset menetelmät ja rajoitukset
Molekyyliorbitaalit perustuvat kvanttimekaniikkaan; käytännön laskennassa sovelletaan erilaisia approksimaatioita. LCAO on yleinen lähestymistapa, ja sen päälle rakennetaan menetelmiä kuten Hartree‑Fock, Density Functional Theory (DFT) tai monielektronimenetelmät (post‑Hartree‑Fock), jotka ottavat huomioon elektronikorrelaation eri tavoin. Laskutoimituksissa käytetään myös basis‑joukkoja (esim. STO‑3G, 6‑31G*), jotka määrittelevät, millaisista funktioista MOit koostetaan.
MO‑mallit tarjoavat usein selkeän ja matemaattisesti käsiteltävän kuvan, mutta niillä on rajoituksensa: hyvin korreloituneissa järjestelmissä yksinkertainen Hartree‑Fock ei riitä, ja lokaalit kemialliset ilmiöt voivat joskus hahmottua paremmin valence bond ‑käsitteellä. Usein paras käytäntö on yhdistää eri lähestymistapoja ja vertailla laskennallisia tuloksia kokeelliseen dataan.
Käytännön havaintoja ja visualisointi
Tietokoneet pystyvät nykyään laskemaan MOita ja visualisoimaan ne kolmiulotteisina pintamuotoina; tällaiset kuvat näyttävät, missä elektronit todennäköisesti sijaitsevat ja miten orbitaalit lomittuvat. MO‑kuvat ja energiadia‑grammit auttavat keskustelemaan sidosten vahvuudesta, elektronisiirtymistä (esim. UV‑VIS‑spektrit) ja reaktiivisuudesta (HOMO/LUMO‑tulkinta).
Yhteenvetona: molekyyliorbitaalit ovat kvanttimekaaninen työkalu, joka yhdistää atomiorbitaalit ja selittää monia molekyylien sähköisiä ja kemiallisia ominaisuuksia. Ne tarjoavat selkeän tavan ymmärtää sidoksia, elektronien jakautumista ja reaktioalttiutta, kun käytetään sopivia laskennallisia menetelmiä ja huomioidaan niiden rajoitukset.

Historia
Robert S. Mulliken käytti sanaa orbitaali ensimmäisen kerran englanniksi. Saksalainen fyysikko Erwin Schrödinger kirjoitti MO:ista jo aiemmin. Schrödinger kutsui niitä nimellä Eigenfunktion.
Fyysikko Max Born kuvasi molekyyliorbitaalien teorian vuonna 1926. Nykyään se tunnetaan Bornin sääntönä, ja se on osa kvanttimekaniikan Kööpenhaminan tulkintaa. Kun teoriaa alun perin ehdotettiin, se ei sopinut yhteen Niels Bohrin atomimallin kanssa. Bohrin malli kuvasi elektronien "kiertävän" ydintä, kun ne liikkuivat ympyrää. Bornin malli sai kuitenkin lopulta suuren suosion, koska se pystyi kuvaamaan elektronien sijainnit molekyyleissä ja selitti useita aiemmin selittämättömiä kemiallisia reaktioita.
Yleiskatsaus
Atomiorbitaalit ennustavat elektronin sijainnin atomissa. Molekyyliorbitaalit syntyvät, kun atomiorbitaalit yhdistetään. Molekyyliorbitaali voi antaa tietoa molekyylin elektronikonfiguraatiosta. Elektronikonfiguraatio on yhden (tai yhden elektroniparin) todennäköisin sijainti ja energia. Useimmiten MO esitetään atomiorbitaalien lineaarisena yhdistelmänä (LCAO-MO-menetelmä), erityisesti likimääräisessä käytössä. Tämä tarkoittaa sitä, että kemistit olettavat, että elektronin todennäköisyys olla missä tahansa molekyylin kohdassa on summa todennäköisyyksistä, joilla elektroni on siellä yksittäisten atomiorbitaalien perusteella. LCAO-MO on yksinkertainen malli molekyylien sidoksista, ja se on tärkeä molekyyliorbitaaliteorian tutkimisessa.
Teoreettiset kemistit käyttävät tietokoneita laskeakseen eri molekyylien MO:t (sekä todelliset että kuvitteelliset). Tietokone voi piirtää "pilvestä" kuvaajia, jotka osoittavat, kuinka todennäköisesti elektroni on millä tahansa alueella. Tietokoneet voivat myös antaa tietoa molekyylin fysikaalisista ominaisuuksista. Ne voivat myös kertoa, kuinka paljon energiaa tarvitaan molekyylin muodostamiseen. Tämä auttaa kemistejä sanomaan, voidaanko joitakin pieniä molekyylejä yhdistää suuremmiksi molekyyleiksi.
Useimmat nykyiset tavat tehdä laskennallista kemiaa aloitetaan laskemalla systeemin MO:t. Kunkin MO:n sähkökenttä syntyy kaikkien atomien ytimien ja muiden elektronien jonkin keskimääräisen jakauman perusteella.
Analogia
Toimintatapojen ymmärtäminen on kuin tietäisi, missä kukin työntekijä on suuressa rautakaupassa (katsomatta kaupan sisälle). Analyytikko tietää myymälässä työskentelevien työntekijöiden määrän ja kunkin työntekijän osaston. Hän tietää myös, että työntekijät eivät astu toistensa varpaille ja että työntekijät seisovat pikemminkin käytävillä kuin tavarahyllyillä. Työntekijät poistuvat omalta osastoltaan auttaakseen asiakkaita löytämään tavaraa muilta osastoilta tai tarkistaakseen varaston. Analyytikko, joka antaa kaikkien myymälässä olevien työntekijöiden sijainnin tiettynä hetkenä katsomatta myymälän sisälle, on kuin kemisti, joka laskee molekyylin MO:n. Aivan kuten MO:t eivät voi kertoa kunkin elektronin tarkkaa sijaintia, myöskään jokaisen työntekijän tarkkaa sijaintia ei tiedetä. MO, jolla on solmutaso, on kuin päätelmä siitä, että työntekijät kävelevät käytäviä pitkin eivätkä hyllyjen läpi. Vaikka elektronit tulevat tietystä atomista, elektroni täyttää MO:n ottamatta huomioon lähdeatomiaan. Tämä on kuin työntekijä, joka lähtee osastoltaan kävelemään muualle myymälässä päivän aikana. MO on siis epätäydellinen kuvaus elektronista aivan kuten analyytikon laskelmat näkymättömästä myymälästä ovat epätäydellinen arvaus työntekijöiden sijainnista.

Molekyyliorbitaalien muodostuminen
Teoreettiset kemistit ovat keksineet sääntöjä MO:iden laskemiseksi. Nämä säännöt perustuvat kvanttimekaniikan ymmärtämiseen. Kvanttimekaniikan avulla kemistit voivat käyttää fysiikan elektronista sanomaa selvittääkseen, miten elektronit käyttäytyvät molekyyleissä. Molekyyliorbitaalit muodostuvat atomiorbitaalien välisistä "sallituista" vuorovaikutuksista. (Vuorovaikutukset ovat "sallittuja", jos atomiorbitaalien symmetriat (jotka on määritetty ryhmäteorian avulla) ovat yhteensopivia keskenään). Kemistit tutkivat atomiorbitaalien vuorovaikutuksia. Nämä vuorovaikutukset syntyvät kahden atomiorbitaalin välisestä päällekkäisyydestä (mitta siitä, kuinka hyvin kaksi orbitaalia on rakentavassa vuorovaikutuksessa keskenään). Päällekkäisyys on tärkeää, jos atomiorbitaalit ovat energialtaan lähellä toisiaan. Molekyylin MO:iden lukumäärän on oltava yhtä suuri kuin niiden atomien atomiorbitaalien lukumäärän, jotka yhdistetään molekyylin muodostamiseksi.
Laadullinen lähestymistapa
Kemistien on ymmärrettävä MO:iden geometria, jotta he voivat keskustella molekyylien rakenteesta. LCMO-menetelmä (Linear combination of atomic orbitals molecular orbital) antaa karkean mutta hyvän kuvauksen MO:ista. Tässä menetelmässä molekyyliorbitaalit ilmaistaan molekyylin jokaisen atomin kaikkien atomiorbitaalien lineaarikombinaatioina.
Atomiorbitaalien lineaariset yhdistelmät (LCAO)
Friedrich Hund ja Robert S. Mulliken esittivät molekyyliorbitaalit ensimmäisen kerran vuosina 1927 ja 1928.
Sir John Lennard-Jones esitteli vuonna 1929 atomiorbitaalien lineaarisen yhdistelmän eli LCAO-approksimaation molekyyliorbitaaleille. Hänen uraauurtava artikkelinsa osoitti, miten fluori- ja happimolekyylien elektroninen rakenne voidaan johtaa kvanttimekaniikan periaatteista. Tämä kvalitatiivinen lähestymistapa molekyyliorbitaaliteoriaan on osa modernin kvanttikemian alkua.
Atomiorbitaalien lineaaristen yhdistelmien (LCAO) avulla voidaan arvata molekyyliorbitaalit, jotka muodostuvat, kun molekyylin atomit liittyvät toisiinsa. Samoin kuin atomiorbitaalille, myös molekyyliorbitaalille voidaan muodostaa elektronin käyttäytymistä kuvaava Schrödingerin yhtälö. Atomiorbitaalien lineaariset yhdistelmät (atomien aaltofunktioiden summat ja erotukset) antavat likimääräiset ratkaisut molekyylien Schrodingerin yhtälöihin. Yksinkertaisille kaksiatomisille molekyyleille saadut aaltofunktiot esitetään matemaattisesti yhtälöiden avulla.
Ψ = ca ψa + cb ψb
ja
Ψ* = ca ψa - cb ψb
missä Ψ ja Ψ* ovat molekyylin aaltofunktiot sitoutuville ja sitoutumattomille molekyyliorbitaaleille, ψa ja ψb ovat atomien a ja b aaltofunktiot, ja ca ja cb ovat säädettävät kertoimet. Nämä kertoimet voivat olla positiivisia tai negatiivisia riippuen yksittäisten atomiorbitaalien energioista ja symmetrioista. Kun kaksi atomia lähestyy toisiaan, niiden atomiorbitaalit limittyvät toisiinsa ja muodostavat suuren elektronitiheyden alueita. Kahden atomin välille muodostuu siis molekyyliorbitaaleja. Atomeja pitää yhdessä sähköstaattinen vetovoima positiivisesti varautuneiden ytimien ja negatiivisesti varautuneiden elektronien välillä, jotka ovat sidoksissa olevilla molekyyliorbitaaleilla.
Sitovat, antisitovat ja ei-sitovat MO:t
Kun atomiorbitaalit ovat vuorovaikutuksessa keskenään, syntyvä molekyyliorbitaali voi olla kolmentyyppinen: sitova, antisitova tai ei-sitova.
Sitoutumismuodot:
- Atomiorbitaalien väliset sidosvuorovaikutukset ovat konstruktiivisia (vaiheen sisäisiä) vuorovaikutuksia.
- Sitovat MO:t ovat energialtaan matalampia kuin atomiorbitaalit, jotka yhdistyvät tuottaakseen ne.
Antibonding MOs:
- Atomiorbitaalien väliset antisidonnaiset vuorovaikutukset ovat tuhoavia (vaiheen ulkopuolisia) vuorovaikutuksia.
- Sitoutumattomien MO:iden energia on korkeampi kuin niiden muodostavien atomiorbitaalien energia.
Ei-sitovat MO:t:
- Sitoutumattomat MO:t ovat seurausta siitä, että atomiorbitaalien välillä ei ole vuorovaikutusta yhteensopivien symmetrioiden puuttumisen vuoksi.
- Sitoutumattomilla MO:illa on sama energia kuin molekyylin yhden atomin atomiorbitaaleilla.
HOMO ja LUMO
Jokaisella molekyyliorbitaalilla on oma energiatasonsa. Kemistit lajittelevat MO:t energiatasojen mukaan. Kemistit olettavat, että elektronit täyttävät ensin matalimman energiatason MO:t. Jos esimerkiksi molekyylissä on elektroneja, jotka täyttävät 15 orbitaalia, täytetään ne 15 MO:ta, joiden energiataso on alhaisin. Luettelon 15. MO:ta kutsutaan "korkeimmin miehitetyksi molekyyliorbitaaliksi" (HOMO) ja 16. MO:ta "alimmaksi miehittämättömäksi molekyyliorbitaaliksi" (LUMO). HOMO:n energiatason ja LUMO:n energiatason eroa kutsutaan kaistaväliksi. Bändiaukko voi joskus toimia molekyylin herätettävyyden mittarina: mitä pienempi energia, sitä helpommin se herää. Kun elektroni herätetään, se hyppää miehittämättömään MO:hon. Tämän avulla voidaan esimerkiksi arvioida, lähettääkö jokin aine valoa (luminesenssi).

Kysymyksiä ja vastauksia
Q: Mikä on molekyyliorbitaali?
A: Molekyyliorbitaali (tai MO) on matemaattinen funktio, joka kuvaa elektronin aaltomaista käyttäytymistä molekyylissä. Se selittää, mitä tapahtuu elektroneille, kun atomit liittyvät yhteen molekyylissä, ja se voi kertoa todennäköisyyden elektronin löytymiselle joltakin tietylle alueelle.
K: Miten kemistit rakentavat matemaattisia molekyyliorbitaalimalleja?
V: Kemistit rakentavat yleensä matemaattisia molekyyliorbitaalimalleja yhdistelemällä atomiorbitaaleja. Myös molekyylin kunkin atomin hybridiorbitaaleja tai muita atomiryhmien molekyyliorbitaaleja voidaan käyttää. Tietokoneet voivat työstää näitä funktioita.
K: Miten kvanttimekaniikka liittyy molekyylien tutkimiseen?
V: Molekyyliorbitaalien avulla kemistit voivat soveltaa kvanttimekaniikkaa molekyylien tutkimiseen. Ne vastaavat kysymyksiin siitä, miten molekyylien atomit pysyvät yhdessä, ja antavat tietoa kemiallisista ja fysikaalisista ominaisuuksista.
K: Mitä ovat orbitaalikaaviot?
V: Orbitaalikaaviot ovat visuaalisia esityksiä, jotka osoittavat, missä atomissa elektronit todennäköisimmin sijaitsevat sen eri pyöreiden muotojen perusteella.
K: Miten hybridiorbitaalit toimivat?
V: Hybridiorbitaalit yhdistävät erityyppisiä atomiorbitaaleja yhdeksi uudeksi tyypiksi, jolla on ainutlaatuisia ominaisuuksia verrattuna sen osiin. Näitä hybridejä käytetään usein, kun rakennetaan matemaattisia molekyyliorbitaalien malleja.
K: Miten tietokoneet voivat auttaa MO:iden tutkimisessa?
V: Tietokoneet voivat auttaa MO:iden tutkimisessa työstämällä niiden toimintoja ja antamalla tarkempia ennusteita tai selityksiä molekyylien kemiallisille ja fysikaalisille ominaisuuksille.
Aiheeseen liittyvät artikkelit
Tekijä
AlegsaOnline.com Molekyyliorbitaali – määritelmä, muodostuminen ja kvanttimekaniikka Leandro Alegsa
URL: https://fi.alegsaonline.com/art/65861
Lähteet
- nobelprize.org : Nobelprize.org