Creutzfeldt–Jakobin tauti (CJD): prionien aiheuttama neurodegeneraatio
Creutzfeldt–Jakobin tauti (CJD) — harvinainen prionien aiheuttama nopea neurodegeneraatio: oireet, diagnostiikka ja ennuste kuolemaan johtavasta sairaudesta.
Creutzfeldt-Jakobin tauti (lausutaan KROITS-felt YAH-kohb) eli CJD on neurologinen sairaus. Se on degeneratiivinen (pahenee ajan myötä), ei parannettavissa ja lopulta aina kohtalokas. CJD:tä kutsutaan joskus "hullun lehmän taudin" (naudan spongiformisen enkefalopatian eli BSE:n) ihmismuodoksi. BSE liittyy varsinaiseen tautimuotoon, joka aiheuttaa yhden harvinaisen Creutzfeldt-Jakobin taudin tyypin; nämä kaksi eivät ole sama tauti mutta ne voivat olla yhteydessä toisiinsa altistuksen kautta.
CJD:n aiheuttaa tartunnanaiheuttaja, jota kutsutaan prioniksi. Prionit ovat proteiineja, jotka ovat taittuneet väärin. Väärin taittuneet prioniproteiinit pystyvät muuttamaan normaalisti taittuneita proteiineja epänormaaliksi muodoksi ja näin levittämään poikkeavan rakenteensa: ne "kopioivat" itseään ilman perinteistä DNA- tai RNA–välitystä. CJD aiheuttaa sen, että aivokudos meni nopeasti epäterveeseen tilaan; tauti tuhoaa aivojen soluja ja aivoihin syntyy mikroskooppisia reikiä, minkä seurauksena aivojen rakenne muuttuu ja niistä tulee spongiainen eli kuin keittiösieni.
Kuvagalleria
6 Kuvat




Oireet ja kulku
CJD:n tyypillinen oireisto etenee nopeasti viikoista kuukausiin. Tavallisimpia oireita ovat:
- nopea etenevä muistin heikkeneminen ja dementia
- kouristelut ja myoklonukset (nopeat lihaskrampit)
- näköhäiriöt ja näköaistin heikkeneminen
- ataxia eli koordinaation ja tasapainon häiriöt
- käytösmuutokset, sekavuus ja unihäiriöt
Sairauden kesto on yleensä lyhyt verrattuna moniin muihin dementiaan aiheuttaviin sairauksiin: monet potilaat menehtyvät muutamien kuukausien tai vuoden sisällä oireiden alkamisesta. Poikkeuksiakin esiintyy, ja harvoissa tapauksissa kulku voi olla hitaampi.
Tauti- ja tautityypit
- Sporadinen CJD – yleisin muoto (noin 85–90 %), jonka syytä ei yleensä tiedetä. Tyypillinen aloitusikä on noin 60 vuotta.
- Familiaalinen (perinnöllinen) CJD – johtuu PRNP-geenissä olevista mutaatioista ja kattaa noin 10–15 % tapauksista.
- Iatrogeeninen CJD – harvinainen, johtuu lääketieteellisestä tartunnasta, esimerkiksi epäasianmukaisesti desinfioiduista instrumenteista tai saastuneista kasvutekijöistä aiemmin käytettyjen kudosvalmisteiden kautta.
- Variant (vCJD) – yhteydessä BSE-epidemiaan; vaikuttaa useammin nuorempiin ihmisiin ja liittyy todennäköisesti altistukseen saastuneelle naudanlihatuotteelle.
Diagnoosi
Diagnoosi perustuu oireisiin, aivokuvantamiseen ja laboratorioeläin- tai -testituloksiin. Käytettyjä tutkimuksia ovat:
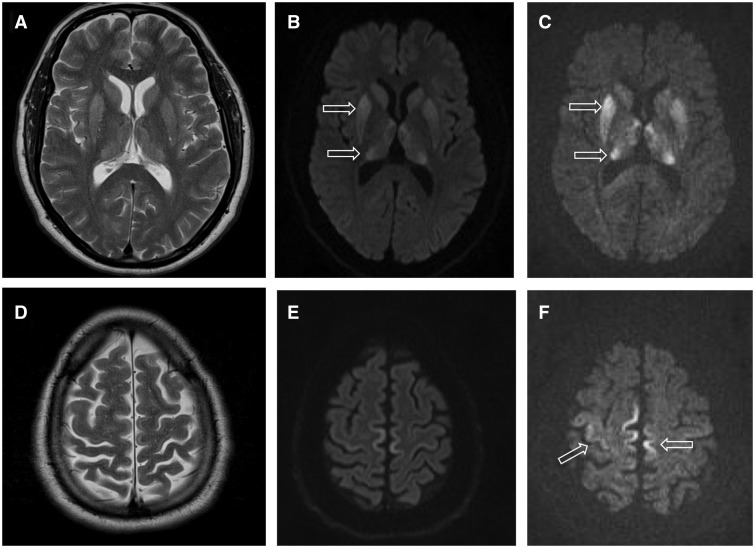
- magnettikuvaus (MRI) – voi näyttää tyypillisiä muutoksia tyvitumakkeissa ja aivokorteksissa ("cortical ribboning")
- EEG – sporadisessa CJD:ssä voi näkyä toistuvia periodisia teräväsäröisiä komplekseja
- likvoritutkimukset – 14-3-3-proteiinin tai korkean totaalitau-arvon nousu voi tukea diagnoosia
- RT-QuIC (real-time quaking-induced conversion) – herkkä ja spesifinen testi prionien tunnistukseen likvorista tai muista kudosnäytteistä
- PRNP-geenitestit – jos epäillään perinnöllistä muotoa
- biopsia tai post mortem -tutkimus – varmistava tutkimus aivediagnostisesti on neuropatologinen tutkimus
Levintätavat ja ehkäisy
Prionitaudit leviävät eri tavoilla riippuen tyypistä. Sporadinen CJD ei ole osoitettu leviävän tavallisessa arkikontaktissa. Variant CJD on yhdistetty ravinnon kautta tapahtuneeseen altistukseen BSE:n yhteydessä. Iatrogeeninen tartunta on mahdollinen terveydenhuollossa, jos instrumentteja tai kudoksia ei desinfioida tai käsitellä asianmukaisesti.
Prioneja on vaikea inaktivoida; ne kestävät tavallisia desinfektioita ja lämpökäsittelyjä. Terveydenhuollossa käytetään erityisiä varotoimia: kertakäyttöisiä välineitä, erityisiä puhdistustoimenpiteitä ja standardeja, jotka sisältävät voimakkaiden emästen tai hypokloritin käyttöä ja korkeapainehöyrysterilisointia pidennetyillä ohjelmilla. Nämä toimenpiteet perustuvat kansainvälisiin ja kansallisiin ohjeisiin.
Hoito ja ennuste
Tällä hetkellä CJD:hen ei ole olemassa parantavaa hoitoa. Hoito on pääosin oireenmukaista ja tukevan hoidon tavoitteena on lievittää oireita ja parantaa potilaan ja läheisten elämänlaatua. Tutkimus uusista hoidoista on käynnissä, mutta toistaiseksi tulokset eivät ole johtaneet tehokkaisiin lääkehoitoihin.
Ennuste on huono: useimmat potilaat menehtyvät vuoden sisällä oireiden alkamisesta, keskimääräinen elinaika on muutamasta kuukaudesta noin vuoteen riippuen sairauden tyypistä ja yksilöllisistä tekijöistä.
Epidemiologia
CJD on harvinainen: sporadisen CJD:n esiintyvyys on suunnilleen 1–2 tapausta miljoonaa ihmistä kohti vuodessa. Variant- ja iatrogeeniset muodot ovat selvästi harvinaisempia.
Yhteenveto
CJD on harvinainen, prionien aiheuttama neurodegeneratiivinen sairaus, joka etenee nopeasti ja on kohtalokas. Se voi esiintyä spontaanisti, perinnöllisesti tai harvoin tartunnan kautta. Diagnoosi perustuu kliinisiin oireisiin, kuvantamistutkimuksiin ja prionitesteihin. Koska parantavaa hoitoa ei ole, korostuvat varhainen tunnistaminen, oireiden hoito ja tartuntaketjujen estäminen terveydenhuollossa.
CJD:n tyypit ja syyt
CJD:n tyyppejä ovat:
- variantti (vCJD):
Tämä CJD-tyyppi voi aiheutua syömällä ruokaa, jossa on prioneja, kuten BSE-tautia sairastavien lehmien lihaa ("hullun lehmän tauti"). Tämä on kuitenkin hyvin harvinainen CJD:n syy.
- sporadinen (sCJD):
Tämä on yleisin CJD-tyyppi. 85 prosenttia CJD-tapauksista on sporadista CJD:tä. Kukaan ei tiedä, mikä aiheuttaa sCJD:n; se näyttää tapahtuvan satunnaisesti.
- familiaalinen (fCJD):
Suurin osa muista 15 prosentista CJD-tapauksista on familiaalista CJD:tä. Tämä on CJD:n muoto, joka esiintyy suvussa.
- iatrogeeninen:
Tämä CJD:n muoto aiheutuu yleensä lääketieteellisestä toimenpiteestä, jossa henkilö saa verta tai kudosta henkilöltä, jolla on CJD. Henkilö voi esimerkiksi saada iatrogeenisen CJD:n, jos hän saa verensiirron tai sarveiskalvonsiirron henkilöltä, jolla on CJD.
Merkit ja oireet
CJD:n ensimmäinen oire on dementia, joka pahenee hyvin nopeasti. dementia aiheuttaa muistin menetystä, persoonallisuuden muutoksia ja aistiharhoja.
Muita yleisiä psyykkisiä oireita ovat:
CJD:n fyysisiä oireita ovat usein:
- Ongelmia puhumisessa
- Tärisevät liikkeet (myoklonus)
- Tasapainohäiriöt (ataksia)
- Kävelyvaikeudet
- Vapina tai jäykkyys
- Näköongelmat
- Nielemisvaikeudet, jotka voivat tehdä syömisestä vaikeaa tai mahdotonta.
- Yskimisvaikeudet, jotka voivat aiheuttaa keuhkokuumetta.
- Liikkeet, joita potilas ei pysty hallitsemaan (dyskinesia).
Useimmat CJD:hen sairastuneet kuolevat kuuden kuukauden kuluessa ensimmäisten oireiden ilmaantumisesta. Usein he kuolevat keuhkokuumeeseen, joka johtuu yskimisvaikeuksista. Noin 15 prosenttia potilaista selviää hengissä kaksi tai useampia vuosia. Jotkut potilaat ovat eläneet 4-5 vuotta enimmäkseen psyykkisin oirein, kunnes tauti pahenee ja aiheuttaa enemmän fyysisiä oireita. Kun näin tapahtuu, ihmiset kuolevat yleensä vuoden kuluessa.
CJD:n oireet johtuvat siitä, että yhä useammat aivojen hermosolut kuolevat. Kun tutkijat tarkastelevat CJD-potilaan aivokudosta mikroskoopilla, he näkevät monia pieniä reikiä, joissa kokonaisia hermosolualueita on kuollut.
Diagnoosi
Lääkärit voivat epäillä CJD:tä, kun henkilöllä on tiettyjä oireita. Esimerkiksi dementia pahenee yleensä hitaasti. Hyvin nopeasti paheneva dementia on epätavallista. Yhdessä nykivien liikkeiden kaltaisten oireiden kanssa nämä oireet voivat viitata mahdolliseen CJD:hen.
Tämän jälkeen voidaan tehdä testejä, joilla voidaan osoittaa, onko henkilöllä CJD. Näihin testeihin kuuluvat:
- Sähköenkefalografia (EEG): Tämä testi osoittaa aivojen sähköisen toiminnan. Lääkäri pystyy usein havaitsemaan EEG:ssä muutoksia, jotka ovat yleisiä CJD:tä sairastavilla. EEG:ssä näkyvät muutokset riippuvat siitä, minkä tyyppistä CJD:tä potilas sairastaa ja kuinka pitkälle sairaus on edennyt.
- Lumbaalipunktio (selkäydinpunktio): Tämä testi mahdollistaa aivo-selkäydinnesteen (aivoja ja selkäydintä ympäröivä neste) tutkimisen tietyn proteiinin ("14-3-3-proteiini") etsimiseksi.
- Aivojen magneettikuvaus: Testi, jossa käytetään erittäin voimakasta magneettia aivojen kuvaamiseen.
- Biopsia: Biopsian ottamiseksi kirurgi ottaa neulalla pienen kudospalan kehosta, jotta lääkäri voi tarkastella sitä mikroskoopilla. vCJD voidaan diagnosoida nielurisojen biopsian avulla. Kaikkien muiden CJD-tyyppien osalta aivobiopsia on ainoa tapa todeta varmasti, onko henkilöllä CJD. Koska aivobiopsia voi kuitenkin aiheuttaa aivovaurion, aivobiopsiaa ei yleensä tehdä, jos muut testit ovat jo osoittaneet, että henkilöllä on todennäköisesti CJD.

Hoito
Vuoteen 2016 mennessä ei ole olemassa hoitoa, joka parantaisi CJD:n tai edes hidastaisi sen vaikutuksia. Hoitomuotojen löytämiseksi tehdään monia kokeita.
Nykyään CJD:n ainoat hoidot ovat lääkkeitä, jotka hoitavat taudin oireita ja auttavat potilaita viihtymään paremmin. Esimerkiksi potilaille, joilla on kouristuksia, voidaan antaa kouristuslääkkeitä. Bentsodiatsepiinit voivat saada lihasnykäykset harvemmiksi.
Potilaat voivat myös valita lääketieteellisiä toimenpiteitä pahojen oireiden lievittämiseksi. CJD voi esimerkiksi aiheuttaa niin suuria nielemisvaikeuksia, ettei henkilö pysty syömään. Joillekin CJD:tä sairastaville laitetaan syöttöletku, kun he eivät enää pysty syömään. Tämä on putki, joka menee vatsaan, jolloin erityistä nestettä voidaan antaa suoraan vatsaan ravinnon antamiseksi henkilölle.
Aiheeseen liittyvät sivut
- Prioni
- Prionisairaus
- Kuolemaan johtava sairaus
Kysymyksiä ja vastauksia
K: Mikä on Creutzfeldt-Jakobin tauti?
V: Creutzfeldt-Jakobin tauti (CJD) on neurologinen sairaus, joka on rappeuttava, parantumaton ja aina kuolemaan johtava.
K: Onko CJD:hen olemassa parannuskeinoa?
V: Ei, CJD:hen ei ole parannuskeinoa.
K: Miksi CJD:tä kutsutaan joskus "hullun lehmän taudin" ihmismuodoksi?
V: CJD:tä kutsutaan joskus "hullun lehmän taudin" ihmismuodoksi, koska naudan spongiforminen enkefalopatia (BSE), joka on yhden harvinaisen CJD:n tyypin aiheuttaja, tunnetaan yleisesti nimellä "hullun lehmän tauti".
K: Mikä on CJD:n syy?
V: CJD:n aiheuttaa tartunnanaiheuttaja, jota kutsutaan prioniksi, joka on proteiini, joka on taittunut väärin ja joka voi tehdä itsestään kopioita muuttamalla oikein taittuneita proteiineja väärin taittuneiksi.
K: Mitä aivokudokselle tapahtuu CJD:ssä?
V: CJD aiheuttaa sen, että aivokudos muuttuu hyvin nopeasti epäterveeksi, jolloin aivoihin syntyy reikiä ja aivojen rakenne muuttuu keittiösienen kaltaiseksi.
K: Onko BSE sama tauti kuin CJD?
V: Ei, BSE ei ole sama tauti kuin CJD; se on itse asiassa yhden harvinaisen CJD-tyypin syy.
K: Miten prionit aiheuttavat CJD:n?
V: Prionit aiheuttavat CJD:tä taittumalla väärin ja tekemällä itsestään kopioita aivoissa olevien oikein taittuneiden proteiinien kustannuksella. Tämä johtaa terveen aivokudoksen tuhoutumiseen ja taudille ominaisten reikien kehittymiseen.
Aiheeseen liittyvät artikkelit
Tekijä
AlegsaOnline.com Creutzfeldt–Jakobin tauti (CJD): prionien aiheuttama neurodegeneraatio Leandro Alegsa
URL: https://fi.alegsaonline.com/art/24152
Lähteet
- cdc.gov : "CJD (Creutzfeldt–Jakob Disease, Classic)"
- ncbi.nlm.nih.gov : "Creutzfeldt–Jakob disease: Transmissible spongiform encephalopathy; vCJD; CJD; Jacob-Creutzfeldt disease"
- bmj.com : "Bovine spongiform encephalopathy and variant Creutzfeldt–Jakob disease"
- doi.org : 10.1101/SQB.1996.061.01.052
- pubmed.ncbi.nlm.nih.gov : 9246478
- www3.interscience.wiley.com : "MM2-cortical-type sporadic Creutzfeldt–Jakob disease with early stage cerebral cortical pathology presenting with a rapidly progressive clinical course"
- doi.org : 10.1111/j.1440-1789.2008.00904.x
- pubmed.ncbi.nlm.nih.gov : 18410280
- who.int : who.int: "Fact sheets no 180: Variant Creutzfeldt-Jakob disease" Feb 2012 ed.
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- doi.org : 10.1056/NEJM198610163151605
- pubmed.ncbi.nlm.nih.gov : 3762620
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- merckmanuals.com : "Creutzfeldt–Jakob Disease (CJD)"
- ncbi.nlm.nih.gov : "Creutzfeldt-Jakob disease: Updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy"